

背景介紹
低溫嚴重損害了鋰離子電池的性能,因此鋰離子電池需要具有寬流動性范圍、離子擴散速度快和脫溶劑化能較低的電解質。而這里面的關鍵在于在Li+和溶劑分子之間建立溫和的內部相互作用,這在商業碳酸亞乙酯基電解質中很難實現。
正文部分
成果簡介
近日,湖南大學劉繼磊教授,構建了低介電常數(ε)溶劑主導配位的溶劑化結構,并通過羰基氧的電負性調節碳酸亞乙酯的配位強度。改性電解質在?90 °C時表現出高離子電導率(1.46 mS·cm?1)?,并在?110 °C時保持液態。因此,4.5?V石墨基軟包電池在?10 °C的200次循環中達到約98%的容量?保持率,沒有鋰枝晶。這些電池在?70?°C時也能保持約60%的室溫放電容量,即使在約?100?°C的溫度下也能奇跡般地保持放電功能。該研究以題目為“Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery”的論文發表在國際頂級期刊《Nature Communications》上。

圖文導讀
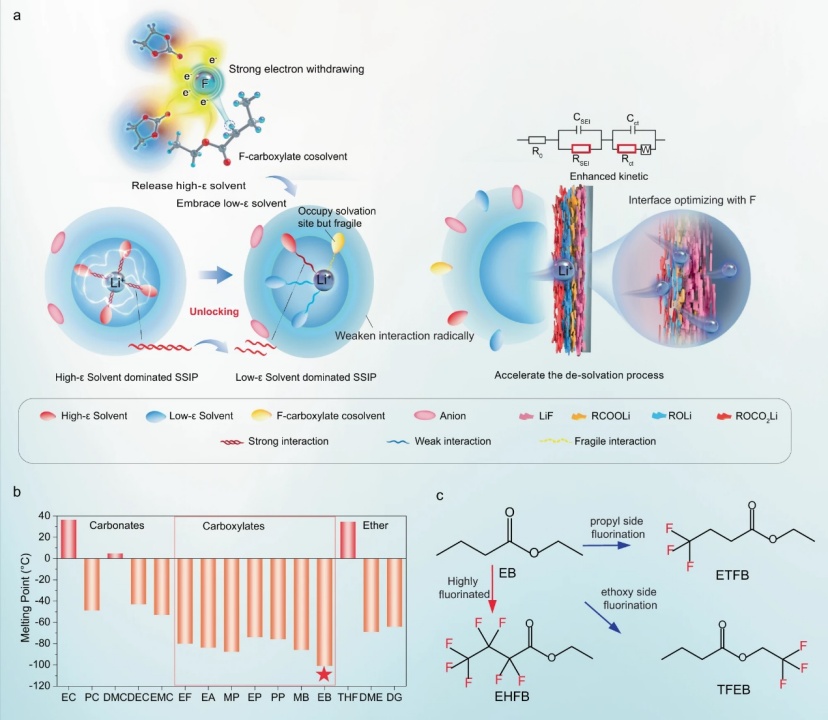
【圖1】a低溫電解質設計原則。b常見碳酸鹽和羧酸鹽溶劑的熔點。c所選EB及其氟化類似物共溶劑的化學結構。
在LIB電解質中,Li+通常通過電負性羰基氧與碳酸鹽電解質中的極性溶劑分子配位。理論上,在不犧牲極性溶劑的高ε性質的情況下削弱這種配位的一種方法是降低高ε溶劑中羰基氧的電負性,而不是用低ε溶劑取代它。這可能通過引入強吸電子元素來實現,例如氟。氟化預計會削弱高ε溶劑(即環狀碳酸酯)和Li+之間的配位相互作用(圖1a),導致(i)釋放更多的高ε溶劑,這將促進低ε溶劑(如線性碳酸酯)與Li+的配位,占據其配位位點,從而將溶劑分離的離子對(SSIP)從高ε溶劑為主轉化為低ε溶劑為主,以及(ii)打破剩余配位的高ε溶劑的相互作用(圖1a)。獨特的溶劑化結構具有幾個優點:(i)通過降低高ε溶劑的配位數和強度以及促進低ε溶劑主導的溶劑化結構的形成,顯著而徹底地削弱了Li+和溶劑之間的整體配位相互作用(圖1a)。此外,氟化羧酸鹽共溶劑也將通過羰基參與溶劑化,即使其與Li+的相互作用減弱(圖1a)。所有這些變化一起促進了所需溶劑化結構的形成,其中Li+和所有溶劑之間的相互作用弱得多,這有望拓寬液體溫度范圍和進一步促進Li+脫溶劑化。(ii)由于較低的LUMO能量,氟化共溶劑將有利于富F SEI的形成。此外,氟化會降低電解質的HOMO能量,從而拓寬其在高壓電池中的電化學窗口。這種方法提供了一種更有效的方法來恢復所需的溶劑化結構,并有望將其應用擴展到極低的溫度。基于這些考慮,本工作將丁酸乙酯(EB)確定為要氟化的溶劑,因為其熔點低(圖1b)。開發了一系列含有EB類似物作為共溶劑的高性能LIB電解質,包括具有不同氟化程度或氟位點的4,4,4-三氟丁酸乙酯(ETFB)、七氟丁酸乙酯和2,2,2-三氟丁酸乙基酯(TFEB)(圖1c)。

【圖2】a 靜電勢(ESP)圖顯示了本工作中考慮的溶劑分子電荷分布。b Li+與不同溶劑的結合能。c基礎電解質和含有EB、ETFB、EHFB和TFEB的電解質電導率。d上述電解質的DSC曲線。e不同電解質在?110 °C下儲存后的圖像,持續30?min。f溶劑化結構與電解質物理性質的關系。
氟化EB衍生物在羰基O原子周圍的電子密度低于EC或EB(圖2a),導致與Li+的結合較弱(圖2b)。此外,氟化程度影響助溶劑的電負性和Li+-溶劑配位強度(圖2b)。例如,EHFB-Li+在含氟共溶劑中具有最低的結合能(~1.72?eV),并且在混合溶劑中EHFB系統的EC-Li+具有最低的結合能(0.71?eV)。這些結果表明,氟化不僅降低了丁酸乙酯本身的配位,而且削弱了周圍EC和Li+之間的相互作用,后者可以用“偶極-偶極效應”來解釋。具體地說,氟的強電子親和力導致電荷分布從C=O端偏移到-CF3端,這改變了氟化丁酸乙酯和周圍EC分子之間的偶極-偶極相互作用,最終導致EC對Li+的弱溶劑化。
弱溶劑化結構顯著改善了電解質的物理性質(圖2f),如離子電導率和流動性。這是因為(i)一些EC分子從溶劑化結構中釋放,以及(ii)更多低熔點的DEC和共溶劑參與溶劑化結構,導致較低的熔點(圖2d)和較低的體相電解質粘度(圖2c插圖)。具體而言,估計冰點增加的順序為EHFB(?135?°C),TFEB(?132?°C)和ETFB(?130?°C)(圖2d),遵循與EC-Li+結合能相同的趨勢。EHFB電解質即使在?110?°C下儲存30分鐘依然能保持液態(圖2e)。基于粘度效應和介電效應(圖2f),高離子電導率取決于的高介電常數和低粘度,因此具有高介電常數和低粘度的EHFB電解質在?90?°C下實現了約1.46 mS·cm?1的高離子電導率?(圖2c)。相反,基礎電解質在約?50?°C時凍結(圖2d),當溫度降至?90?°C時,顯示出較差的離子導電性(0.001 mS·cm?1)(圖2c)。

【圖3】基于分子動力學(MD)模擬計算出?70°C下基礎電解質(a)和EHFB電解質(b)的徑向分布函數(g(r))和配位數(n(r)。c 在EHFB電解質中不同溫度下EC和DEC的FTIR光譜C=O峰區域。d用Voigt函數擬合?70?°C下EHFB電解質的FTIR光譜。基礎電解質(e)和EHFB電解質(f)中EC的對稱環變形和(C–O)鍵的拉伸振動拉曼光譜。g基于計算的RDF,五種電解質在25?°C時的配位數。h 25?°C和?70?°C下基礎電解質和EHFB電解質中所有溶劑組分與Li+的配位數。i在不同溫度下,基礎電解質和EHFB電解質中的溶劑化EC與游離EC以及溶劑化EC與溶劑化DEC的比值。J當溫度從25°C降低至?70?°C時,基礎電解質和EHFB電解質中溶劑化結構變化示意圖?。
通過分子動力學(MD)模擬將溶劑化結構解耦,并提供了在每種電解質情況下計算的Li+的徑向分布函數(RDF)(g(r))和配位數(n(r))(圖3a、b)。每種溶劑的第一溶劑化半徑為~2.65??,PF6?占據第二個溶劑化殼層(~4.2??)形成分離的離子對(SIP-PF6?)。在基礎電解質中,EC是第一Li+溶劑化殼中的主溶劑,室溫下的配位數為1.31,然后是線性碳酸鹽DEC(1.13)和EMC(0.94)(圖3g)。然而,隨著上述共溶劑的加入,Li+溶劑化結構發生了顯著變化。具體而言,EHFB電解質中的EC配位數急劇下降至0.61(圖3b和圖3g),而DEC配位數上升至1.46(圖3b和圖3g)。在EB、ETFB和TFEB系統中觀察到類似的趨勢,表明從EC主導的溶劑化結構轉變為DEC主導的溶劑化結構。這些轉變與氟化相關,并且共溶劑的氟化度越高,DEC溶劑與Li+的配位就越強,EC與Li+之間的相互作用就越弱。這些影響在較低的溫度下更為明顯。例如,EHFB電解質中的DEC配位數從25°C時的1.46增加至?70?°C時的1.71,而EC配位數從0.61下降到0.33(圖3h),表明在LT的第一個溶劑化殼中,更多的EC被DEC取代。相反,在基礎電解質中,隨著溫度的降低,EC與Li+的配位數增加(從25°C時的1.34增加到-70°C時的1.76),而DEC配位數減少(從25°C時的1.13減少到-70°C時的0.74)。考慮到配位每個Li+的溶劑分子總數恒定,EC配位的減少不可避免地導致DEC配位的增加。顯然,減少EC配位的優勢不會被DEC配位的增加所掩蓋,這歸因于DEC相對于EC固有的低極性性質。一方面,由于EC是一種高極性溶劑,因此EC配位數的減少無疑會導致溶劑化的減弱。另一方面,DEC的配位數的增加也導致溶劑化的減弱,因為其極性較低。因此,這兩個因素協同促進了整體弱化溶劑化結構的形成。這突出了EHFB共溶劑在LT應用中的優點。除了氟化共溶劑對主要溶劑溶劑化的影響外,它們還影響Li+溶劑化,因為EB、ETFB、TFEB和EHFB的配位數隨著氟化的增加從1.3、0.85、0.74降至0.06(圖3g)。這與計算的Li+與溶劑結合能十分一致(圖2b)。此外,EHFB的配位數從25?°C時的0.06急劇增加到?70?°C時的0.56(圖3h)。這表明EHFB在LT下對Li+的溶劑化更具活性。低ε溶劑與共溶劑一起主導了溶劑化結構,導致Li+與內溶劑化鞘中所有溶劑之間的相互作用減弱(圖3j)。這些特征促進了Li+的脫溶劑化,從而增強了LT下的電化學動力學。
溫度相關的FTIR和拉曼光譜分析進一步證實了共溶劑與主溶劑一起對Li+溶劑化的貢獻(圖3c-f)。游離和溶劑化C=O的IR峰(在1660-1870 cm?1的區域內)用Voigt函數擬合(圖3d)。溶劑化EC與游離EC之比(R1)和溶劑化EC與溶劑化DEC之比(R2)(等式1和等式2)用于量化溶劑化EC的相對豐度以及EC和DEC之間的配位競爭。
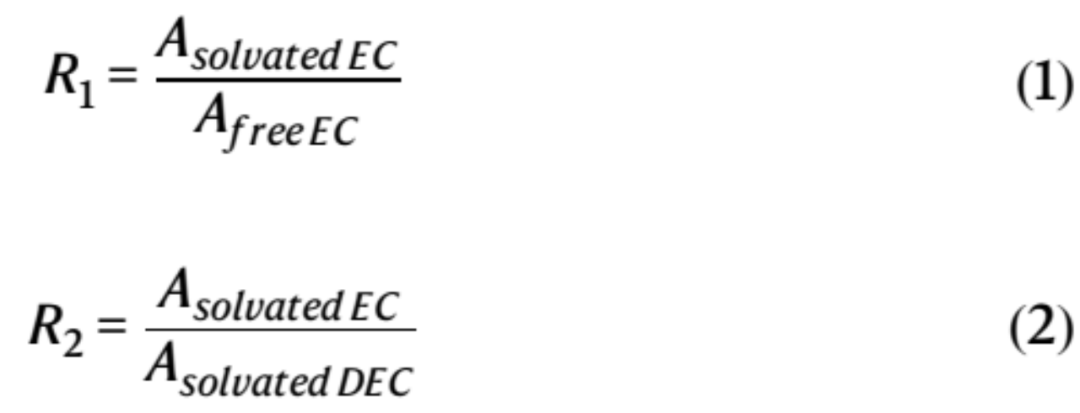
其中Asolvated EC、Afree EC和Asolvate DEC是溶劑化EC、游離EC和溶劑化DEC的C=O基團對應的振動峰積分面積強度(圖3d)。定量分析顯示,隨著溫度從25℃下降至?70?°C,R1和R2值都有所下降(圖3i)。例如,EHFB電解質的R1值從1.34(25?°C)下降至1.09(?70?°C),同時R2從1.53(25?°C)下降至0.66(?70?°C)(圖3i)。這表明,共溶劑促進了DEC和Li+之間的配位,同時EC和Li+間的相互作用減弱,尤其是在LT。注意,與ETFB和TFEB相比,EHFB電解質的R2值下降得更快(圖3i),表明其在LT下對DEC主導的溶劑化結構有更大的選擇性。相反,對于基礎電解質,R1和R2都隨著溫度從25?°C降低至?70?°C而增加,證實了在寬溫范圍內EC主導的溶劑化作用(圖3i)。此外,這種變化伴隨著拉曼光譜中兩個溶劑化EC峰的顯著藍移(從741到749?cm?1,從904到911?cm?1)(圖3e),進一步表明在LT基礎電解質中,Li+和EC之間的配位相互作用更強。這降低了其在寒冷條件下的實用性。這些特征,包括降低的R1和R2值(圖3i)和抑制的C-O峰藍移(圖3f),證明共溶劑能夠減弱EC的配位強度,從而設計了以DEC為主溶劑,共溶劑參與的溶劑化結構,最終形成了弱溶劑化結構(圖3j)。
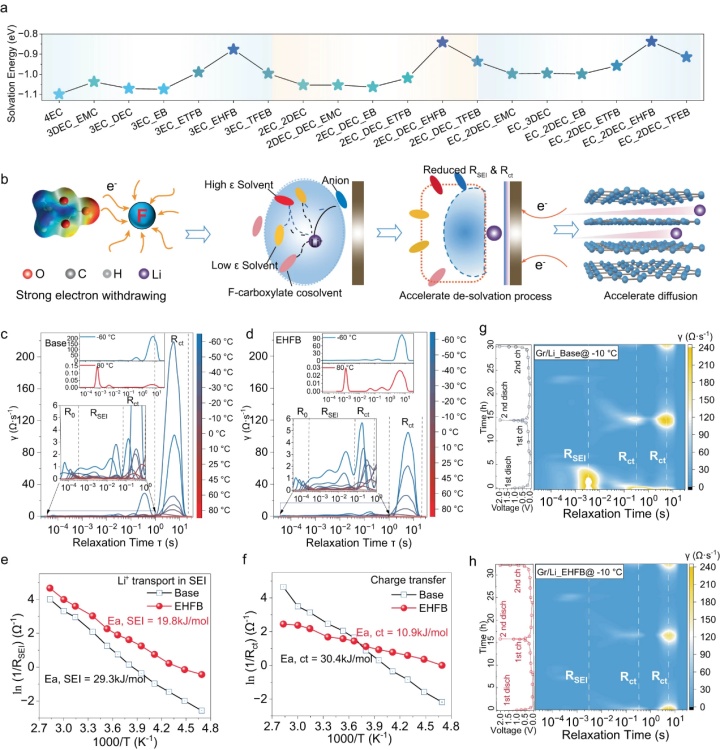
【圖4】a不同電解質中的Li+溶劑化/脫溶劑化能。b氟化共溶劑在削弱Li+和溶劑分子之間的相互作用、加速脫溶劑化和促進Li+擴散方面的關鍵作用。根據采用基礎電解質(c)和EHFB電解質(d)的LCO/Gr軟包電池EIS數據得出的弛豫時間分布(DRT)的溫度依賴性圖。通過Arrhenius擬合Rct(e)和RSEI(f)得到相應的活化能。?10?°C下,采用基礎電解質(g)和EHFB電解質(h)的石墨/Li半電池兩個循環的原位DRT數據。
DFT計算表明,氟化共溶劑顯著降低了EC/DEC電解質的Li+脫溶劑化能(圖4a),并且這一趨勢高度依賴于氟化。共溶劑的氟化程度越高,脫溶劑化能越小(從4EC的1.1?eV至EC-2DEC-EB的1.0? eV,EC-2DEC-ETFB的0.93? eV和EC-2DEC-EHFB的0.83? eV)。這是由于氟取代基的強吸電子效應,增強了Li+和低ε溶劑之間的配位,并形成了DEC主導的、總體減弱的溶劑化結構,從而促進了脫溶劑化過程(圖4b)。
通過對弛豫時間(DRT)分布分析進一步驗證了氟化共溶劑對Li+脫溶劑化的影響(圖4c,d)。該分析通過連續分布函數中的局部最大值對不同的電化學過程進行了分類。Rct和RSEI都表現出強烈的溫度依賴性。低溫時Rct占主導地位,高溫時RSEI占主導地位。具體而言,EHFB電解質在?60°C時具有1.0 Ω的最低Rct(圖4d),然后是TFEB(3.2Ω)和ETFB(4.7Ω)。這些值遠小于基礎電解質(8.8Ω)(圖4c),表明助溶劑在促進電荷轉移中的重要作用。電荷轉移的活化能較低也證實了這一點(圖4f),并且具有EHFB電解質的LCO/Gr軟包電池活化能估計為10.9?kJ·mol?1,約為基礎電解質的三分之一(30.4?kJ·mol?1)。活化能按EHFB<TFEB<?ETFB的順序增加,與脫溶劑化能的趨勢非常一致(圖4a)。采用基礎電解質和EHFB電解質的Gr/Li半電池在?10°C下兩次電化學循環的DRT圖譜(圖4g,h)表明,EHFB系統表現出比基礎電解質小得多的Rct,證實了共溶劑的引入促進了電荷轉移,特別是在低溫運行的石墨負極中。此外,與基礎電解質(29.3?kJ·mol?1)(圖4e)相比,具有EHFB的全電池中Li+通過SEI的活化能低得多(19.8?kJ·mol?1)(圖4f),因此RSEI顯著降低(圖4g,h)。此外,與基礎電解質不同(圖4g),EHFB系統中的RSEI略有變化,其值要小得多(圖4h),這表明源自EHFB的SEI具有高導電性,并且比基礎電解質中的SEI更堅固。
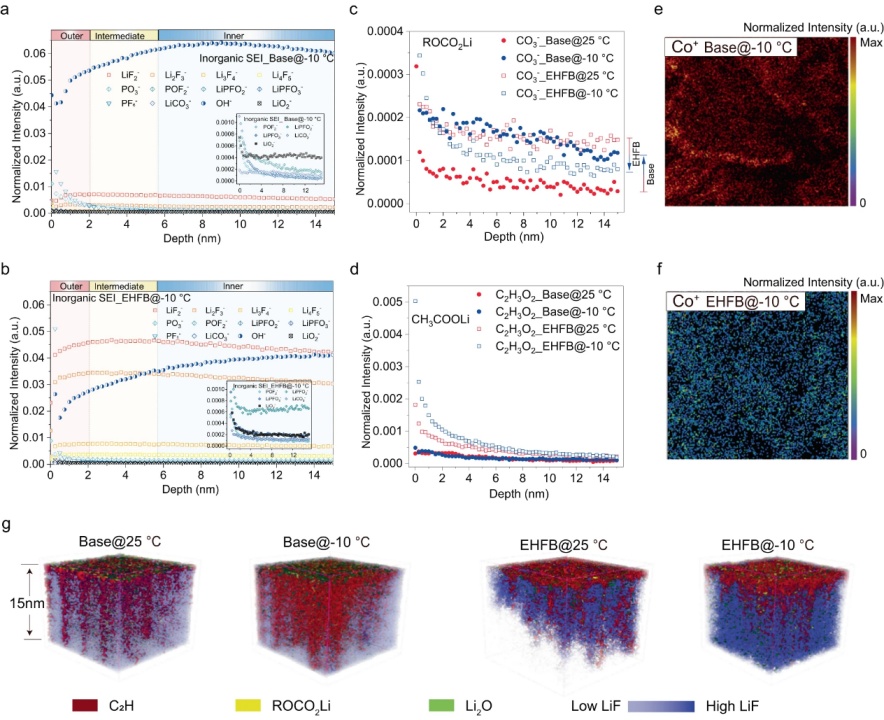
【圖5】在基礎電解質(a)和EHFB電解質(b)中,?10°C下長循環后無機SEI中官能團的深度分布。25 °C和?10?°C時基礎電解質和EHFB電解質形成的SEI中ROCO2Li組分(c)和CH3COOLi組分的深度分布?。采用基礎電解質(e)和EHFB電解質(f)的石墨負極上Co+的空間分布。g在25°C和-10°C下,采用基礎電解質和EHFB電解質的電池化成后石墨負極SEI最上面15nm的三維重建圖。
用飛行時間二次離子質譜法(TOF-SIMS)表征了循環石墨電極上SEI層的成分/結構演變。深度剖面TOF-SIMS數據顯示,有機部分(CH2?、CO3?、C2H3O?和C2H3O2?)主要集中在SEI的外表面(圖5c、d),而無機LiF物種(LiF2?、Li2F3?、Li3F4?、Li4F5?)和OH?在SEI內側更普遍(圖5a、b)。離子碎片潛在來源包括三種主要有機成分:ROCO2Li(CO3?碎片)、CH3COLi(C2H3O?)和CH3COOLi(C2H3O2?),它們分別通過EC、DEC和羧酸鹽的電化學還原產生。在基礎電解質中,ROCO2Li信號隨著溫度從25?°C降低至?10?°C而增強(圖5c),而CH3COLi信號減少,并且幾乎沒有檢測到CH3COOLi信號(圖5d)。這表明在基礎電解質中EC是LT下進行還原的主要溶劑,而不是DEC。相反,?10?°C下,當使用EHFB電解質時,ROCO2Li信號降低,CH3COLi信號增加(圖5c),表明在EHFB中,LT增強了DEC還原并抑制了EC還原。在基礎電解質中,EC對Li+的溶劑化在LT時得到促進,但在EHFB情況下得到緩解。此外,檢測到的CH3COOLi信號表明,EHFB共溶劑參與LT時石墨表面的成膜(圖5d)。此外,在源自EHFB電解質的界面中檢測到更多的LiF物種(圖5a,b),并且它們的量隨著溫度的降低而增加。SEI性質的差異反映在不同溫度下有機SEI組分(C2H,ROCO2Li)和無機物種(LiF,Li2O)的3D空間分布覆蓋圖中(圖5g)。具體而言,在EHFB中,僅在SEI的最外層中發現少量有機組分(即C2H),并且LT下更多的LiF存在于SEI內層中。這可能是由于更多的EHFB參與溶劑化并有助于LT時SEI的形成。相反,基礎電解質衍生的SEI中LiF少得多(圖5a,圖5d)。此外,在EHFB電解質中,RT和LT條件下Co+的溶解和串擾被有效抑制(圖5e、f)。這可能是由于氟化電解質的抗氧化性,用它可以很好地保護正極結構在約4.5 V的高電壓下不受損壞。這突出了氟化共溶劑在高壓應用中的顯著優勢和SEI的優異性能。相反,在基礎電解質中,大量的Co+被溶解,然后串擾到石墨負極,這對SEI具有高度破壞性(圖5e、f)。
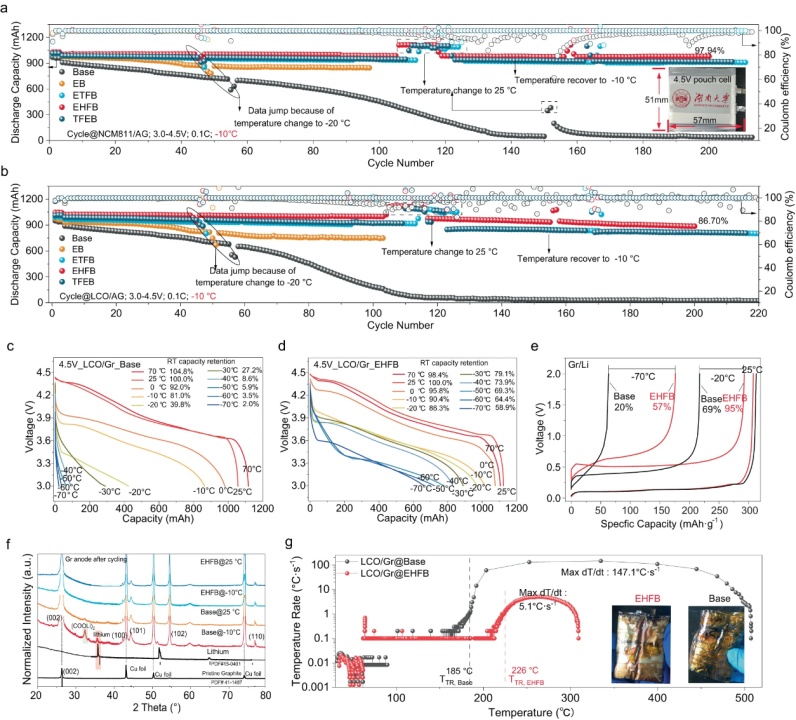
【圖6】NCM811/Gr(a)和LCO/Gr(b)電池在?10°C下不同電解質中的循環行為。具有基礎電解質(c)和EHFB電解質(d)的LCO/Gr軟包電池RT充電-LT放電的電壓曲線。e具有不同電解質的Gr/Li電池在不同溫度下的充電電壓曲線。f在?10°C和25?°C下長循環后石墨的XRD圖譜。g完全充電LCO/Gr電池的溫升速率隨溫度的變化。插圖顯示了測試后的照片。
將EHFB電解質與商業1Ah NCM811/Gr和LCO/Gr軟包電池匹配獲得了優異的低溫電化學性能。在?10?°C下200次循環后,NCM811/Gr的容量保持率為97.94%,LCO/Gr的容量保持率為86.70%(圖6a,b)。利用RT充電-LT放電方案,使用EHFB電解質的LCO/Gr軟包電池在?40 °C時保持830 mAh的高容量?,相當于其RT容量的73.9%。即使在?70°C時,這些電池仍保留了約60%的RT容量(圖6d)。相反,使用基礎電解質的LCO/Gr和NCM811/Gr電池表現出較差的性能,在?10 °C時僅保留約80%的RT容量?,在?40 °C時完全失效?(圖6c)。電池的低溫性能受到石墨負極的限制,這是由鋰枝晶引起的。事實證明,當在?10 °C下循環時,石墨負極基電池表現出顯著的容量衰減?(圖6a,b)。因此,對Gr/Li電池在低溫下的性能進行了評估。采用EHFB電解液的Gr/Li電池在-70°C和-20°C下充電,室溫放電后分別保持了57%和95%的容量。這些值遠高于基礎電解質的值(分別為20%和69%的RT容量,圖6e)。XRD分析顯示,在?10°C循環的石墨負極中,在35.8°處觀察到明顯的鋰峰(圖6f),而在EHFB情況下不存在。這些發現都證實了改性電池在低溫下能有效抑制鋰枝晶的形成。使用加速量熱法(ARC)評估安全性能(圖6g)。結果表明,與基礎電解質相比,使用EHFB電解質的LCO/Gr軟包電池安全性顯著提高。EHFB電解質導致最大dT/dt顯著降低,僅為5.1?°C s?1和高得多的熱失控溫度(TTR,226?°C),能夠有效避免熱失控事件。此外,最高溫度降至309?°C,表明熱失控過程中釋放的總能量大幅減少。
總結和展望
本工作展示了一種使用傳統EC基電解質的溶劑化設計策略,以提高LIBs低溫性能。通過引入氟化共溶劑,削弱了EC與Li+的強配位,并將EC主導的溶劑化結構轉變為DEC主導的溶劑化結構,特別是在低溫下。這種設計促進了Li+的脫溶劑化,同時保持了EC的高介電性能。此外,氟化共溶劑也有助于形成富氟SEI,增強了石墨負極在寒冷條件下的穩定性。因此,在低溫性能方面取得了顯著的改進,例如更寬的流動性范圍(將液體保持在?110?°C),電導率更高(在?90 °C時為1.46 mS·cm?1?),更容易的脫溶劑化過程,并抑制了鋰枝晶的生長。這使得1 Ah 4.5?V石墨基軟包電池在?10?°C下穩定循環200多次,當在?60?°C下充放電一圈時,只有2%的容量損失并保持334 mAh的容量。此外,電池在?70°C時放電?,仍有60%的室溫容量,并能夠在約?100 °C的極低溫度下為設備供電?。這項工作為開發適用于極端環境的鋰離子電池提供了一種獨特的方法。
參考文獻
Yuqing Chen, Qiu He, Yun Zhao, Wang Zhou, Peitao Xiao, Peng Gao, Naser Tavajohi, Jian Tu, Baohua Li, Xiangming He, Lidan Xing, Xiulin Fan & Jilei Liu*. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery, Nature Communications.
DOI:10.1038/s41467-023-43163-9
https://doi.org/10.1038/s41467-023-43163-9
注:圖片非商業用途,存在侵權告知刪除!
本文地址:http://www.lbzrq0002.com/news/details1886.html
好文章,需要你的鼓勵
郵箱:libatterychina@163.com
北京:北京市海淀區上地三街9號金隅嘉華大廈C座904
010-62980511
山東:山東省臨沂市魯商中心A12-1503-1
0539-8601323
鋰電中國(libattery.net)版權所有
Copyright By 北京貝特互創科技有限公司
京ICP備11002324號-1
京公安網備11010802035676號

 手機鋰電網
手機鋰電網





我有話說: